I have a GLMM with a binomial distribution and a logit link function and I have the feeling that an important aspect of the data is not well represented in the model.
To test this, I would like to know whether or not the data is well described by a linear function on the logit scale. Hence, I would like to know whether the residuals are well-behaved. However, I can not find out at which residuals plot to plot and how to interpret the plot.
Note that I am using the new version of lme4 (the development version from GitHub):
packageVersion("lme4")
## [1] ‘1.1.0’
My question is: How do I inspect and interpret the residuals of a binomial generalized linear mixed models with a logit link function?
The following data represents only 17% of my real data, but fitting already takes around 30 seconds on my machine, so I leave it like this:
require(lme4)
options(contrasts=c('contr.sum', 'contr.poly'))
dat <- read.table("http://pastebin.com/raw.php?i=vRy66Bif")
dat$V1 <- factor(dat$V1)
m1 <- glmer(true ~ distance*(consequent+direction+dist)^2 + (direction+dist|V1), dat, family = binomial)
The simplest plot (?plot.merMod) produces the following:
plot(m1)
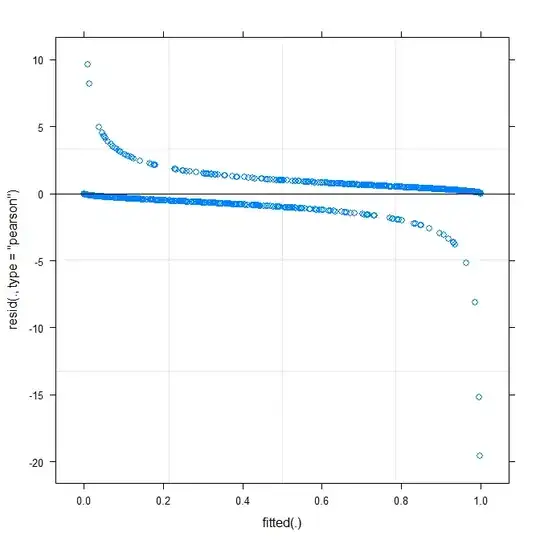
Does this already tell me something?