I am reviewing the R package OpenMx for a genetic epidemiology analysis in order to learn how to specify and fit SEM models. I am new to this so bear with me. I am following the example on page 59 of the OpenMx User Guide. Here they draw the following conceptual model:
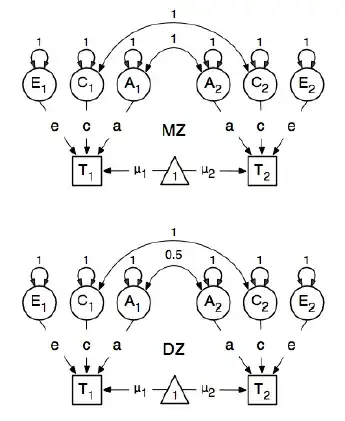
And in specifying the paths, they set the weight of the latent "one" node to the manifested bmi nodes "T1" and "T2" to be 0.6 because:
The main paths of interest are those from each of the latent variables to the respective observed variable. These are also estimated (thus all are set free), get a start value of 0.6 and appropriate labels.
# path coefficients for twin 1
mxPath(
from=c("A1","C1","E1"),
to="bmi1",
arrows=1,
free=TRUE,
values=0.6,
label=c("a","c","e")
),
# path coefficients for twin 2
mxPath(
from=c("A2","C2","E2"),
to="bmi2",
arrows=1,
free=TRUE,
values=0.6,
label=c("a","c","e")
),
The value of 0.6 comes from the estimated covariance of bmi1 and bmi2 (of strictly monozygotic twin pairs). I have two questions:
When they say that the path is given a "starting" value of 0.6 is this like setting a numerical integration routine with initial values, like in estimation of GLMs?
Why is this value estimated strictly from the monozygotic twins?